Teo đa hệ thống (MSA) là một bệnh thoái hóa thần kinh tiến triển tăng dần, thường gặp thứ hai trong hội chứng Parkinson không điển hình. Bệnh biểu hiện với các triệu chứng của hội chứng Parkinson, thất điều tiểu não, rối loạn thần kinh thực vật và hội chứng tháp.
Bài viết “Tổng quan bệnh teo đa hệ thống” trích nguồn từ Hội Thần Kinh học Việt Nam.
Tác giả: Lê Thị Ngọc1, Nguyễn Thanh Bình2
Bệnh viện Lão khoa Trung ương1
Đại học Y Hà Nội2
1. Lịch sử
Năm 1900, Dejerine và Thomas lần đầu tiên mô tả các trường hợp lâm sàng về bệnh teo trám cầu tiểu não. Sau này, đây được xem là một thể của teo đa hệ thống.
Năm 1969, thuật ngữ “teo đa hệ thống” được đưa ra bao gồm cả ba hội chứng lâm sàng là thoái hóa thể vân chất đen, teo trám cầu tiểu não và hội chứng Shy-Drager. Năm 1999, thuật ngữ teo đa hệ thống thể Parkinson với triệu chứng Parkinson chiếm ưu thế (MSA-P) đã được sử dụng thay thế cho thoái hóa thể vân chất đen, teo đa hệ thống thể tiểu não với triệu chứng tiểu não chiếm ưu thế (MSA-C) thay thế teo trám cầu tiểu não. Hội chứng Shy-Drager được sử dụng khi bệnh nhân có triệu chứng thần kinh thực vật chiếm ưu thế.
2. Dịch tễ
Tỷ lệ mắc trung bình hàng năm ước tính là 0,6 đến 0,7 trên 100.000 người. Tỷ lệ hiện mắc ước tính là 3,4 đến 4,9 trường hợp trên 100.000 dân số, tăng lên 7,8 trên 100.000 ở người trên 40 tuổi [1]. Bệnh ảnh hưởng đến cả 2 giới và khởi phát ở khoảng 55 tuổi. Ở hầu hết các quốc gia, teo đa hệ thống thể Parkinson nhiều hơn thể tiểu não tuy nhiên ở Nhật Bản thể tiểu não lại thường gặp hơn.
Nguyên nhân của teo đa hệ thống vẫn chưa rõ ràng, chưa có yếu tố môi trường nào được coi là yếu tố nguy cơ của bệnh. Một số yếu tố liên quan đến môi trường nghề nghiệp như tiếp xúc với kim loại, nhựa, dung môi hữu cơ và thuốc trừ sâu được mô tả trên bệnh nhân teo đa hệ thống, tuy nhiên chưa có bằng chứng cho thấy mối liên quan giữa các yếu tố này và bệnh.
Trong những năm gần đây, các nghiên cứu về di truyền trên bệnh nhân teo đa hệ thống cho thấy sự hiện diện của di truyền trội hoặc lặn trên NST thường. Các đột biến trong gen coenzyme Q2 (gen cần thiết cho quá trình sinh tổng hợp coenzyme Q10) đã được phát hiện ở bệnh nhân MSA. Ngoài ra, người ta cũng nhận thấy mối liên quan của các biến thể gen SNCA, MATP và tăng nguy cơ mắc bệnh teo đa hệ thống. Gen LRRK2 và gen GBA liên quan đến bệnh Parkinson cũng được phát hiện ở bệnh nhân teo đa hệ thống [2].
3. Giải phẫu bệnh
Giải phẫu bệnh lý đặc trưng của teo đa hệ thống là mất tế bào thần kinh và thần kinh đệm trong thể vân, chất đen, nhân xanh, nhân cầu não, cuống tiểu não giữa, tế bào Purkinje tiểu não, nhân olive dưới; cột trung gian, nhân Onuf của tủy sống.
Người ta nhận thấy có sự xuất hiện của α -synuclein trong tế bào chất của tế bào thần kinh đệm oligodendrocytes (hay còn được gọi là các thể vùi) phân bố điển hình ở tiểu não, cầu não và hạch nền. Teo đa hệ thống cùng với bệnh Parkinson và sa sút trí tuệ thể Lewy được chứng minh có chung bản chất là rối loạn của α -synuclein, và được gọi là bệnh α -synuclein.
4. Lâm sàng
Các đặc điểm lâm sàng của teo đa hệ thống bao gồm các triệu chứng vận động và ngoài vận động ở nhiều dạng phối hợp, khác nhau ở từng giai đoạn và từng thể bệnh.
Giống như bệnh Parkinson, teo đa hệ thống có giai đoạn tiền vận động gặp trong 20 – 75% trường hợp, bao gồm rối loạn chức năng tình dục, tiểu không tự chủ hoặc bí tiểu, hạ huyết áp tư thế, thở rít và rối loạn hành vi trong giấc ngủ giai đoạn động mắt nhanh (REM) xuất hiện từ vài tháng đến nhiều năm trước khi các triệu chứng vận động đầu tiên xuất hiện.
4.1. Các triệu chứng vận động
4.1.1. Teo đa hệ thống với hội chứng Parkinson chiếm ưu thế
MSA-P biểu hiện hội chứng Parkinson với những đặc điểm như giảm động, cứng, mất ổn định tư thế và/hoặc run. Run khi nghỉ xảy ra ở 1/3 bệnh nhân MSA-P và các triệu chứng về vận động thường đối xứng. Các dấu hiệu cảnh báo khác của teo đa hệ thống bao gồm mất ổn định tư thế và té ngã (thường trong vòng ba năm sau khi bắt đầu các triệu chứng vận động), các dấu hiệu tháp (dấu hiệu Babinski) và tiến triển nhanh dù đã điều trị dopaminergic [3]. Đáp ứng kém với levodopa là tiêu chuẩn chẩn đoán bắt buộc đối với bệnh teo đa hệ thống. Có 40% bệnh nhân teo đa hệ thống đáp ứng thoáng qua với levodopa trong giai đoạn đầu của bệnh. Các rối loạn vận động khác có thể gặp như giật cơ, múa vung nửa người, múa giật, loạn trương lực cơ. Một số kiểu loạn trương lực cơ như loạn trương lực cơ vùng miệng mặt giống như “cười co thắt” (co thắt cơ mặt gây biểu hiện cười méo mó) và hội chứng Pisa (loạn trương lực cơ bán cấp với sự uốn cong nặng của thân, đầu và cổ sang bên). Ngoài ra, loạn trương lực cơ trục thân – Camptocormia (cột sống gập về phía trước nghiêm trọng) và loạn trương lực cơ cổ gập trước là những bất thường tư thế thường gặp.
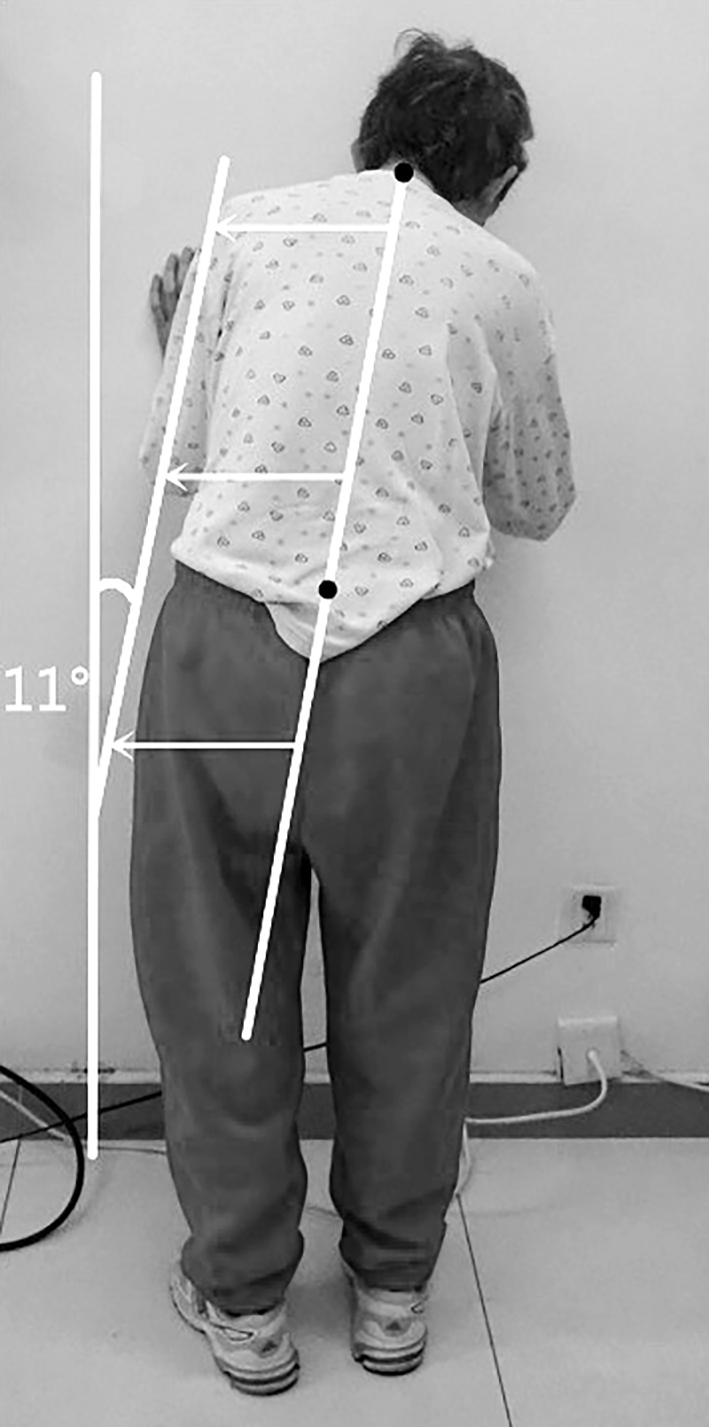
Hình 1. Hội chứng Pisa

Hình 2. Loạn trương lực cơ trục thân – Camptocormia
4.1.2 Teo đa hệ thống với triệu chứng tiểu não chiếm ưu thế
MSA-C chủ yếu biểu hiện các triệu chứng như thất điều dáng đi, thất điều chi, rối loạn vận ngôn và rối loạn chuyển động của mắt. Các bất thường về mắt có thể bao gồm rung giật nhãn cầu, suy giảm khả năng theo đuổi nhịp nhàng, giảm phản xạ tiền đình mắt.
Ở giai đoạn tiến triển của bệnh thường gặp tình trạng ngã tái phát, khó phát âm (thay đổi tông giọng), khó nói (khó nói thành lời), chảy nước dãi và khó nuốt.
4.2 Các triệu chứng ngoài vận động
Rối loạn thần kinh tự chủ
Rối loạn thần kinh thực vật sớm và nặng là đặc điểm chính của teo đa hệ thống. Trong đó triệu chứng tiết niệu sinh dục và tim mạch bị ảnh hưởng thường xuyên nhất.
Nam giới bị teo đa hệ thống thường bị rối loạn cương dương sớm. Rối loạn chức năng tiểu tiện bao gồm tiểu khó, tiểu đêm, tiểu gấp, tiểu dầm tuy nhiên ít khi gặp bí tiểu. Các triệu chứng này thường xuất hiện sớm hơn và nặng hơn trong teo đa hệ thống, trong khi bệnh Parkinson thường gặp ở giai đoạn muộn.
Trong triệu chứng về tim mạch, ha huyết áp tư thế là đặc điểm lâm sàng thường gặp nhất. Ngoài ra, teo đa hệ thống còn có thể có hạ huyết áp sau bữa ăn và hạ huyết áp về đêm. Hiện tượng Raynaud (phản ứng mạch máu quá mức với nhiệt độ lạnh hoặc căng thẳng về cảm xúc) hoặc đầu chi lạnh cũng có thể xuất hiện ở bệnh nhân teo đa hệ thống.
Rối loạn hô hấp
50% bệnh nhân teo đa hệ thống có triệu chứng thở rít ban ngày hoặc ban đêm và xuất hiện thường xuyên hơn ở giai đoạn bệnh nặng. Ngoài ra, bệnh nhân teo đa hệ thống còn xuất hiện ngừng thở khi ngủ do tắc nghẽn (15% đến 37% trường hợp), ngưng thở khi ngủ trung ương, giảm thông khí khi ngủ có thể dẫn đến suy hô hấp cấp tính và đột tử [4].
Rối loạn giấc ngủ
Rối loạn hành vi trong giấc ngủ giai đoạn động mắt nhanh (RBD) gặp ở ít nhất 2/3 bệnh nhân teo đa hệ thống. Bệnh nhân thường có những giấc mơ sống động, bạo lực hoặc đáng sợ. Bệnh nhân có thể nói hoặc la hét trong khi ngủ và có thể tấn công người ngủ cùng. Trong một số trường hợp RBD có thể xảy ra trước các biểu hiện vận động của teo đa hệ thống vài năm hoặc thậm chí hàng chục năm. Buồn ngủ ban ngày quá mức và hội chứng chân không yên xuất hiện ở gần 30% bệnh nhân teo đa hệ thống, tỷ lệ tương tự hoặc cao hơn ở bệnh Parkinson [5].
Chức năng nhận thức
Chức năng nhận thức trong teo đa hệ thống có xu hướng được bảo tồn tương đối tốt so với bệnh Parkinson và các hội chứng Parkinson không điển hình khác. Rối loạn chức năng thùy trán kèm theo mất khả năng chú ý đã được báo cáo trong một phần ba số trường hợp teo đa hệ thống. Không kiểm soát được cảm xúc hay hội chứng giả hành tủy (cười hoặc khóc không thích hợp khi không có bối cảnh cảm xúc phù hợp) cũng như các thay đổi hành vi, bao gồm cả trầm cảm, lo lắng, cơn hoảng sợ và ý định tự tử cũng có thể xảy ra [6].
5. Cận lâm sàng
5.1. Các xét nghiệm hình ảnh học
Vị trí tổn thương chủ yếu của teo đa hệ thống là vùng hạch nền, thân não và tiểu não vì vậy cộng hưởng từ (MRI) luôn là xét nghiệm được ưu tiên để đánh giá tổn thương. Cắt lớp vi tính sọ não được chỉ định trong trường hợp bệnh nhân có chống chỉ định với MRI não, giúp loại trừ các tổn thương não để chẩn đoán phân biệt bệnh teo đa hệ thống với các bệnh lý khác.
– MRI não
MRI là một xét nghiệm đáng tin cậy và dễ tiếp cận để phân biệt teo đa hệ thống với bệnh Parkinson, sa sút trí tuệ thể Lewy và liệt trên nhân tiến triển. Các dấu hiệu của teo đa hệ thống có thể nhìn thấy trên MRI 1,5 Tesla thường quy gồm có: tăng tín hiệu viền ngoài của bèo sẫm (dấu hiệu viền bèo sẫm), teo bèo sẫm, giảm tín hiệu bèo sẫm phía sau trên T2W; dấu chữ thập ở cầu não, teo cầu và tiểu não, tăng tín hiệu cuống tiểu não giữa.
Dấu hiệu “hot cross-bun” (dấu hiệu chữ thập) được biết đến nhiều nhất và đại diện cho sự thoái hóa của cầu não và bó cầu tiểu não với sự bảo tồn của ống tủy. Trên T2 axial xuất hiện tăng tín hiệu dạng chữ chữ thập ở cầu não. Mặc dù đây là dấu hiệu nhận biết MSA-C với độ đặc hiệu cao (97%), nhưng độ nhạy của nó chỉ là 50% [7].
Dấu hiệu “viền bèo sẫm” – Tăng tín hiệu viền ngoài bèo sẫm trên hình ảnh T2 * được tìm thấy trong MSA-P và có độ đặc hiệu cao nhất (90%), độ nhạy 72%. Ngoài ra, MRI MSA-P còn thấy teo bèo sẫm trên xung T1 (độ nhạy 83%, độ đặc hiệu 87%), và giảm tín hiệu bèo sẫm (độ nhạy 89%, độ đặc hiệu 70%) .
MSA-C có những thay đổi như tăng tín hiệu của cuống tiểu não giữa (MCP) và teo tiểu não và thân não (đặc biệt là cầu não) (độ nhạy 100%, độ đặc hiệu 82 %) [9].
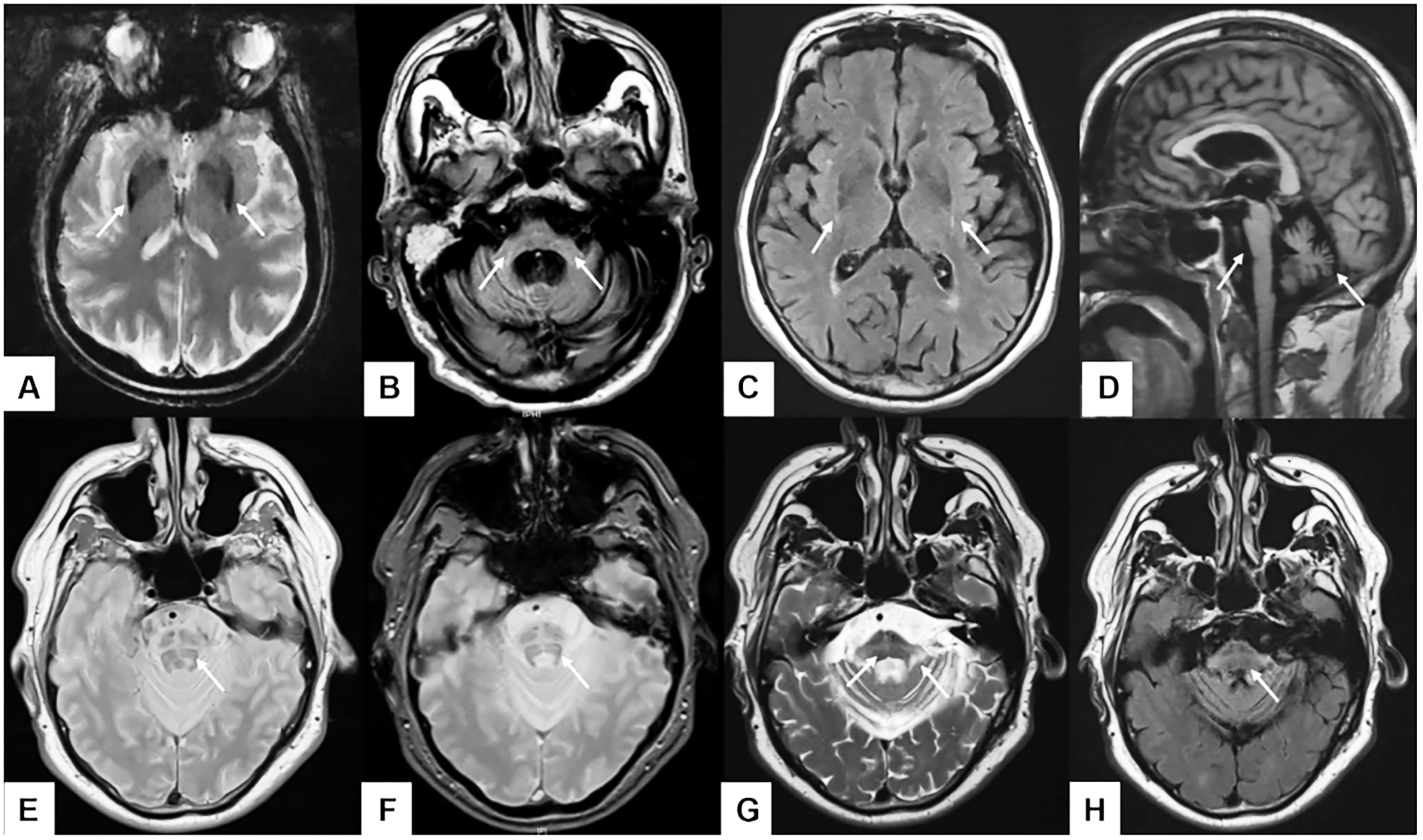
Hình 3. Hình ảnh cộng hưởng từ bệnh nhân teo đa hệ thống:
A: Giảm tín hiệu bèo sẫm, B: teo cuống tiểu não giữa, C: dấu hiệu “viền bèo sẫm”, D: teo cầu não và nhộng tiểu não, E: dấu hiệu chữ thập trên T2 PD W, F: dấu hiệu chữ thập trên T2*, G: dấu hiệu chữ thập và tăng tín hiệu cuống tiểu não giữa trên T2, H: dấu hiệu chữ thập trên T2 FLAIR
Các phương pháp định lượng đơn giản đã được chứng minh là cải thiện độ chính xác của chẩn đoán. Trong teo đa hệ thống teo cầu não ưu thế hơn so với teo não giữa, do vậy người ta sử dụng các phép đo đơn giản đường kính trước sau não giữa, cầu não hay tỷ lệ não giữa/cầu não được đo trên sagittal qua đường giữa để chẩn đoán phân biệt bệnh teo đa hệ thống, bệnh Parkinson và liệt trên nhân tiến triển. Tỷ lệ diện tích não giữa/cầu não ở bệnh nhân MSA-P lớn hơn ở bệnh nhân liệt trên nhân tiến triển. Sử dụng cách đo đường kính trục ngắn, teo đa hệ thống có tỷ lệ não giữa/cầu não > 0.52 [10].
Ngoài ra, đặc điểm teo cuống tiểu não giữa (chiều rộng cuống tiểu não giữa < 8mm ở mặt cắt sagittal) của teo đa hệ thống cũng giúp chẩn đoán phân biệt với bệnh Parkinson có độ nhạy và độ đặc hiệu 100%. Tỷ lệ cuống tiểu não giữa /cuống tiểu não cũng thấp hơn ở bệnh nhân teo đa hệ thống.
Chỉ số Parkinson cộng hưởng từ (MRPI) [= diện tích cầu não /não giữa) * (chiều rộng cuống tiểu não giữa/cuống tiểu não trên)] đang được sử dụng trong các nghiên cứu gần đây có độ đặc hiệu cao hơn nhưng độ nhạy thấp hơn so với tỷ số não giữa/cầu não trong việc chẩn đoán phân biệt teo đa hệ thống và liệt trên nhân tiến triển.

Hình 4. Đo kích thước trước sau cầu não đo ở khoảng cách lớn nhất vuông góc với sàn của não thất tư (A, B), đo kích thước trung não (C, D) [11]
– Chụp cắt lớp phát xạ Positron sử dụng fluorodeoxyglucose 18-F (FDG-PET) ở bệnh nhân MSA có thể thấy tình trạng giảm chuyển hóa glucose khu vực trong thể vân, thân não và tiểu não và mô hình chuyển hóa cụ thể liên quan đến teo đa hệ thống, nhưng độ nhạy và độ đặc hiệu thực sự của những phát hiện này để phân biệt teo đa hệ thống và bệnh Parkinson vẫn chưa chắc chắn.
– SPECT: Chụp cắt lớp vi tính phát xạ đơn photon sử dụng 99m Tc-ECD làm chất đánh dấu cho thấy hình ảnh giảm tưới máu ở thể vân, thân não và tiểu não giúp phân biệt teo đa hệ thống với bệnh Parkinson (độ nhạy 73,3%, độ đặc hiệu 84%).
5.2. Các xét nghiệm khác
– Các xét nghiệm đánh giá rối loạn thần kinh thực vật: holter điện tim và huyết áp 24 giờ giúp đánh giá nguy cơ hạ huyết áp thế đứng.
– Ghi điện cơ co thắt bàng quang
– Siêu âm đánh giá lượng nước tiểu tồn dư
– Ghi đa ký giấc ngủ: chẩn đoán hội chứng ngừng thở khi ngủ, hội chứng chân không yên hoặc các rối loạn hành vi ban đêm.
6. Tiêu chuẩn chẩn đoán
Chẩn đoán chắc chắn bệnh teo đa hệ thống chỉ thực hiện được qua giải phẫu bệnh. Nếu chỉ dựa trên lâm sàng và hình ảnh học, chỉ có thể chẩn đoán teo đa hệ thống ở mức độ “có thể” và “có khả năng”.
Năm 2008, các chuyên gia về teo đa hệ thống (MSA) bao gồm các nhà lâm sàng, bệnh lý học, và hình ảnh học đã thống nhất bản đồng thuận về các tiêu chuẩn chẩn đoán teo đa hệ thống của Hội Thần kinh Hoa Kỳ và Hội Thần kinh Thực vật Hoa Kỳ [12], bao gồm các bảng sau:
Bảng 1: Tiêu chuẩn chẩn đoán “có thể là MSA” (probable MSA):
Bệnh khởi phát ở người trưởng thành trên 30 tuổi, tiến triển dần, xuất hiện rải rác, với các đặc trưng: Rối loạn thần kinh thực vật có liên quan đến tiểu không tự chủ (không có khả năng kiểm soát bài tiết nước tiểu từ bàng quang, nam giới thường xuất hiện cùng rối loạn cương dương) hoặc tụt huyết áp tư thế trong vòng 3 phút khi đứng, với huyết áp tâm thu giảm ít nhất 30mmHg, hoặc huyết áp tâm trương giảm ít nhất 15mmHg, VÀ một trong những đặc điểm sau:
• Hội chứng Parkinson (giảm động với cứng, run, mất thăng bằng tư thế) đáp ứng kém với Levodopa (dưới nhóm Parkinson [MSA-P])
• Hội chứng tiểu não (thất điều dáng đi, loạn vận ngôn do tiểu não – cerebellar dysarthria, thất điều chi, rối loạn chức năng vận nhãn do tiểu não) (dưới nhóm tiểu não [MSA-C])
Bảng 2: Tiêu chuẩn chẩn đoán “có khả năng là MSA” (possible MSA):
Bệnh khởi phát ở người trưởng thành trên 30 tuổi, tiến triển dần, xuất hiện rải rác, với đặc điểm sau:
• Hội chứng Parkinson (giảm động với cứng, run, mất thăng bằng tư thế) HOẶC hội chứng tiểu não (thất điều dáng đi, loạn vận ngôn do tiểu não, thất điều chi, hoặc rối loạn chức năng vận nhãn do tiểu não) VÀ
• Ít nhất 1 đặc điểm gợi ý có rối loạn chức năng thực vật (chứng tiểu gấp mà không giải thích bằng lý do khác, phải tiểu thường xuyên hoặc tiểu không hết bãi, rối loạn cương dương ở nam, hoặc hạ huyết áp tư thế rõ rệt nhưng không thỏa mãn tiêu chí “có thể là MSA”) VÀ
• Ít nhất có 1 dấu hiệu bổ sung, được liệt kê trong bảng 3
Bảng 3: Các dấu hiệu bổ sung của “có khả năng là MSA”:
Có khả năng MSA-P hoặc MSA-C
• Dấu hiệu Babinski kèm tăng phản xạ gân xương.
• Thở rít (stridor).
Có khả năng MSA-P
• Hội chứng Parkinson tiến triển nhanh.
• Đáp ứng kém với levodopa.
• Mất ổn định tư thế trong vòng 3 năm kể từ khi khởi phát triệu chứng vận động.
• Thất điều dáng đi, loạn vận ngôn do tiểu não, thất điều chi, hoặc rối loạn chức năng vận nhãn do tiểu não.
• Khó nuốt trong vòng 5 năm kể từ khi khởi phát triệu chứng vận động.
• Hình ảnh teo các vùng nhân bèo sẫm, cuống tiểu não giữa, cầu não hoặc tiểu não trên phim cộng hưởng từ.
• Giảm chuyển hóa trên phim FDG-PET của bèo sẫm, thân não hoặc tiểu não.
Có khả năng MSA-C
• Hội chứng Parkinson (giảm động và cứng)
• MRI với hình ảnh teo bèo sẫm, cuống tiểu não giữa, cầu não
• Giảm chuyển hóa trên FDG-PET ở bèo sẫm
• Mất phân bố chất dẫn truyền dopamin trước synap tại chất đen – thể vân trên SPECT hoặc PET
Bảng 4: Các đặc điểm hỗ trợ (dấu hiệu cờ đỏ) và không hỗ trợ chẩn đoán MSA
* Đặc điểm hỗ trợ:
• Loạn trương lực cơ miệng mặt
• Loạn trương lực cơ cổ gập trước không cân xứng
• Camptocormia (cột sống gập về phía trước nghiêm trọng) và/hoặc hội chứng Pisa (cột sống cong về một bên nghiêm trọng)
• Co cứng cơ bàn tay hoặc bàn chân
• Thở khò khè
• Rối loạn phát âm nặng
• Khó nói nặng
• Ngáy mới hoặc gia tăng
• Tay và chân lạnh
• Không kiểm soát được cảm xúc (cười hoặc khóc bệnh lý)
• Run giật bất thường hoặc run tư thế hoặc run khi vận động
* Đặc điểm không hỗ trợ chẩn đoán MSA
• Run khi nghỉ kiểu “vê thuốc” điển hình
• Bệnh lý thần kinh ngoại biên rõ rệt trên lâm sàng
• Ảo giác không do thuốc.
• Khởi phát sau 75 tuổi.
• Tiền sử gia đình có biểu hiện thất điều hoặc hội chứng Parkinson.
• Sa sút trí tuệ (phù hợp với tiêu chuẩn DSM-IV).
• Tổn thương chất trắng gợi ý bệnh xơ cứng rải rác.
7. Chẩn đoán phân biệt
Chẩn đoán phân biệt quan trọng của MSA-P là với bệnh Parkinson và với các hội chứng Parkinson không điển hình khác, chủ yếu là liệt trên nhân tiến triển và thoái hóa vỏ hạch nền.
– Bệnh Parkinson:
Trong giai đoạn đầu, khó phân biệt MSA-P với bệnh Parkinson vô căn. Teo đa hệ thống thường đáp ứng kém với levodopa, tiến triển đến tàn tật nhanh chóng và xuất hiện các triệu chứng như rối loạn thần kinh tự chủ rõ, cứng và chậm vận động không tương xứng với run, nói khó nghiêm trọng, thở rít, ngã sớm.
– Liệt trên nhân tiết triển: Bệnh nhân thường có liệt liếc dọc, ngã xuất hiện sớm hơn teo đa hệ thống và không có hạ huyết áp tư thế.
– Thoái hóa vỏ hạch nền: Bệnh nhân thường có mất thực dụng động tác, loạn trương lực cơ, giật cơ, hội chứng bàn tay người ngoài hành tinh, mất cảm giác vỏ não.
MSA-C có thể bị chẩn đoán nhầm với:
– Thất điều tủy tiểu não.
– Thất điều Friendreich khởi phát muộn
– FXTAS: hội chứng run/thất điều có liên quan đến nhiễm sắc thể X dễ gãy
8. Điều trị
Hiện nay chưa có phương pháp điều trị đặc hiệu nào cho teo đa hệ thống, chủ yếu là điều trị triệu chứng, phục hồi chức năng và chăm sóc hỗ trợ.
Triệu chứng vận động
– Levodopa: Các triệu chứng vận động của bệnh nhân teo đa hệ thống thường đáp ứng kém với thuốc. Nên tăng liều levodopa một cách từ từ để giảm thiểu đợt cấp của hạ huyết áp tư thế đứng, phù và buồn nôn. Việc ngừng sử dụng Levodopa đột ngột ở những bệnh nhân không có đáp ứng rõ ràng đôi khi làm nặng hơn các vận động bất thường và không thể hồi phục.
– Thuốc đồng vận dopamine ít có hiệu quả cải thiện vận động và thường làm trầm trọng thêm tình trạng hạ huyết áp thế đứng, các triệu chứng tiêu hóa và rối loạn giấc ngủ trong bệnh teo đa hệ thống.
– Bằng chứng về hiệu quả của amantadine (chất đối kháng chọn lọc các thụ thể NMDA) còn đang tranh cãi.
– Tiêm độc tố botulinum có thể được sử dụng trong trường hợp loạn trương lực cơ ở chi hoặc loạn trương lực cơ trục thân.
– Phục hồi chức năng vận động và ngôn ngữ giúp cho bệnh nhân giảm té ngã và tăng cường khả năng giao tiếp.
Các triệu chứng ngoài vận động
Hạ huyết áp tư thế
Có thể sử dụng fludrocortisone acetate là thuốc điều trị đầu tay để tăng thể tích lòng mạch. Ngoài ra có thể sử dụng midodrine (thuốc chủ vận alpha-1 adrenergic) và droxidopa (tiền chất norepinephrine). Các biện pháp không dùng thuốc bao gồm uống nhiều nước và muối, đi tất áp lực, thay đổi tư thế từ từ và nâng cao đầu giường 10-20 độ.
Trong trường hợp bệnh nhân có hạ huyết áp sau ăn có thể sử dụng octreotide tiêm dưới da 30 phút trước mỗi bữa ăn. Ngoài ra, bệnh nhân nên được tư vấn ăn nhiều bữa nhỏ, các bữa ăn ít carbohydrate, tránh hạn chế muối, tránh đứng dậy đột ngột hoặc đứng yên sau bữa ăn, đi bộ giữa các bữa ăn; nếu đi bộ không được, nằm với đầu giường 45 độ trong 90 phút sau bữa ăn.
Tiết niệu, sinh dục
Bàng quang tăng hoạt gây tiểu không kiểm soát có thể sử dụng các thuốc kháng hệ muscarinic oxybutynin hoặc tolterodine. Đối với bàng quang mất trương lực, sử dụng đặt ống thông ngắt quãng khi thể tích nước tiểu tồn dư >100ml.
Rối loạn cương dương: Các thuốc như sildenafil 50 mg hoặc tadalafil 2,5 đến 5 mg một lần/ngày và các phương pháp vật lý trị liệu khác.
Thở rít
Thở áp lực dương liên tục về đêm là biện pháp điều trị cho hầu hết các bệnh nhân. Mở khí quản trong các trường hợp nặng.
Táo bón
Có thể sử dụng chế độ ăn nhiều chất xơ và chất làm mềm phân; đối với các trường hợp đáp ứng kém có thể cần thụt tháo.
9. Tiến triển và tiên lượng bệnh
Teo đa hệ thống được đặc trưng bởi sự tiến triển xấu đi không ngừng của các triệu chứng vận động và không vận động trong khoảng thời gian trung bình là 10 năm. Khoảng 50% bệnh nhân cần hỗ trợ đi lại trong vòng 3 năm sau khi bắt đầu có các triệu chứng vận động, 60% phải ngồi xe lăn sau 5 năm, và thời gian trung bình trước khi bệnh nhân nằm liệt giường là 6 đến 8 năm. Nguyên nhân tử vong trong bệnh teo đa hệ thống thường bao gồm viêm phế quản phổi, nhiễm trùng tiết niệu, hoặc đột tử. Đột tử thường xảy ra vào ban đêm và được cho là do rối loạn hô hấp trung ương, ngạt thở do đờm và thức ăn, rối loạn thần kinh tự chủ của tim hoặc sự kết hợp của các yếu tố này.
TÀI LIỆU THAM KHẢO
1. Bower J.H., Maraganore D.M., McDonnell S.K. and et al. (1997). Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology, 49(5), 1284–1288.
2. Sailer A., Scholz S.W., Nalls M.A. and et al. (2016). A genome-wide association study in multiple system atrophy. Neurology, 87(15), 1591–1598.
3. Wenning G.K., Colosimo C., Geser F. and et al. (2004). Multiple system atrophy. The Lancet Neurology, 3(2), 93–103.
4. Köllensperger M., Geser F., Seppi K. and et al. (2008). Red flags for multiple system atrophy. Movement disorders: official journal of the Movement Disorder Society, 23(8), 1093–1099.
5. Moreno-López C., Santamaría J., Salamero M. and et al. (2011). Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Archives of neurology, 68(2), 223–230.
6. Stankovic I., Krismer F., Jesic A. and et al. (2014). Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Movement Disorders, 29(7), 857–867.
7. Schrag A., Good C.D., Miszkiel K. and et al. (2000). Differentiation of atypical Parkinsonian syndromes with routine MRI. Neurology, 54(3), 697–697.
8. Lee E.A., Cho H.I., Kim S.S. and et al. (2004). Comparison of magnetic resonance imaging in subtypes of multiple system atrophy. Parkinsonism Relat Disord, 10(6), 363–368.
9. Fanciulli A. và Wenning G.K. (2015). Multiple-system atrophy. New England Journal of Medicine, 372(3), 249–263.
10. Massey L.A., Jäger H.R., Paviour D.C. and et al. (2013). The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology, 80(20), 1856–1861.
11. Warmuth-Metz M., Naumann M., Csoti I. and et al. (2001). Measurement of the Midbrain Diameter on Routine Magnetic Resonance Imaging: A Simple and Accurate Method of Differentiating Between Parkinson Disease and Progressive Supranuclear Palsy. Archives of Neurology, 58(7), 1076–1079.
12. Gilman S., Wenning G.K., Low Pa. al and et al. (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71(9), 670–676.
1. Bower J.H., Maraganore D.M., McDonnell S.K. and et al. (1997). Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology, 49(5), 1284–1288.
2. Sailer A., Scholz S.W., Nalls M.A. and et al. (2016). A genome-wide association study in multiple system atrophy. Neurology, 87(15), 1591–1598.
3. Wenning G.K., Colosimo C., Geser F. and et al. (2004). Multiple system atrophy. The Lancet Neurology, 3(2), 93–103.
4. Köllensperger M., Geser F., Seppi K. and et al. (2008). Red flags for multiple system atrophy. Movement disorders: official journal of the Movement Disorder Society, 23(8), 1093–1099.
5. Moreno-López C., Santamaría J., Salamero M. and et al. (2011). Excessive daytime sleepiness in multiple system atrophy (SLEEMSA study). Archives of neurology, 68(2), 223–230.
6. Stankovic I., Krismer F., Jesic A. and et al. (2014). Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Movement Disorders, 29(7), 857–867.
7. Schrag A., Good C.D., Miszkiel K. and et al. (2000). Differentiation of atypical Parkinsonian syndromes with routine MRI. Neurology, 54(3), 697–697.
8. Lee E.A., Cho H.I., Kim S.S. and et al. (2004). Comparison of magnetic resonance imaging in subtypes of multiple system atrophy. Parkinsonism Relat Disord, 10(6), 363–368.
9. Fanciulli A. và Wenning G.K. (2015). Multiple-system atrophy. New England Journal of Medicine, 372(3), 249–263.
10. Massey L.A., Jäger H.R., Paviour D.C. and et al. (2013). The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology, 80(20), 1856–1861.
11. Warmuth-Metz M., Naumann M., Csoti I. and et al. (2001). Measurement of the Midbrain Diameter on Routine Magnetic Resonance Imaging: A Simple and Accurate Method of Differentiating Between Parkinson Disease and Progressive Supranuclear Palsy. Archives of Neurology, 58(7), 1076–1079.
12. Gilman S., Wenning G.K., Low Pa. al and et al. (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71(9), 670–676.
Leave a Reply