Phình và tách động mạch chủ là một hội chứng di truyền hiếm gặp do đột biến trong gen NF1, gây ra sự phát triển không bình thường của các tế bào thần kinh và các mô liên quan.
1.Hội chứng Marfan
Hội chứng Marfan là một trong những bệnh lý rối loạn mô liên kết di truyền thường gặp nhất trong phình và tách động mạch chủ. Bệnh di truyền trội trên nhiễm sắc thể thường. Hội chứng Marfan liên quan tới sự đột biến của gen FBN1 mã hóa cho fibrillin-1 (thành phần chủ yếu của elastin hoặc elastin liên kết microfibril) qua đó dȁn đến sự tăng phiên mã của yếu tố phát triển và biệt hóa beta (TGF-p).
Dựa theo nghiên cứu của Shore và cộng sự tiến hành năm 1994, thuốc chẹn beta giao cảm trở thành một trong các thuốc điều trị kinh điển của các bệnh nhân có hội chứng Marfan nhằm giảm tiến triển của tổn thương động mạch chủ.
Các nghiên cứu thực nghiệm gần đây cho thấy việc ức chế TGF-beta bằng kháng thể trung hòa hoặc với angiotensin-II Type-1 receptor blocker có thể đảo ngược các biến chứng tại thành mạch. Đây là cơ sở cho việc sử dụng các thuốc ức chế thụ thể (đặc biệt là losartan) trong điều trị hội chứng Marfan.
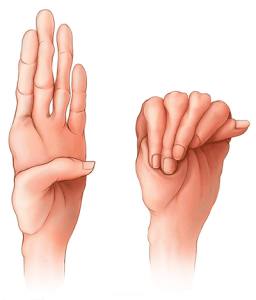
Biểu hiện lâm sàng hội chứng Marfan
2. Hội chứng Turner
Hội chứng Turner gây ra bởi sự thiếu hụt một phần hoặc toàn bộ nhiễm sắc thể X (karotyp 45X0). Chẩn đoán dựa trên triệu chứng lâm sàng và phân tích tế bào học.
Những phụ nữ bị ảnh hưởng của bệnh thường có kiểu hình lùn, các bất thường tim mạch bẩm sinh rất đa dạng, bất thường động mạch chủ, rối loạn hormone và chuyển hóa dȁn đến béo phì, rối loạn dung nạp đường huyết, tăng lipid máu và suy chức năng buồng trứng.
Khoảng 75% bệnh nhân có các bất thường giải phȁu của hệ tim mạch: 12% các bệnh nhân hội chứng Turner có hẹp eo ĐM chủ, 30% có van ĐM chủ có hai lá van. Thường quan sát thấy sự giãn của các động mạch lớn, đáng chú ý nhất là động mạch chủ, động mạch cánh tay và động mạch cảnh. Sự kéo dài của phần quai, giãn ĐM chủ được quan sát thấy với tỷ lệ lần lượt 30% và 33%, phình ĐM chủ thường gặp ở gốc của động mạch chủ lên. Xác định đường kính của động mạch chủ ở người lớn với hội chứng Turner thường khó khăn do không có sự tương xứng giữa tuổi, giới và kích thước cơ thể. Tỷ lệ tách thành động mạch chủ ở phụ nữ mắc hội chứng Turner cao hơn 100 lần so với các phụ nữ bình thường, thường xảy ra ở độ tuổi 30 – 40.
Việc quản lý các bệnh nhân có hội chứng Turner cần dựa trên nguy cơ tim mạch, thăm khám lâm sàng và các phương pháp chẩn đoán hình ảnh. Đối với nhóm bệnh nhân nguy cơ thấp, siêu âm qua thành ngực mỗi 3 – 5 năm. Đối với nhóm nguy cơ trung bình, chụp MRI lồng ngực mỗi 3 – 5 năm. Đối với nhóm nguy cơ cao, cần khám chuyên khoa tim mạch, chụp phim MRI lồng ngực mỗi 1 – 2 năm.
3. Hội chứng Ehlers-Danlos Type IV hay type mạch máu
Là hội chứng rối loạn mô liên kết hiếm gặp trong phình và tách động mạch chủ, di truyền trội nhiễm sắc thể thường do đột biến gen COL3A1, mã hóa cho procollagen Typ III.
Chẩn đoán dựa trên các dấu hiệu lâm sàng, thăm dò hình ảnh không xâm lấn và tình trạng đột biến của gen COL3A1. Đặc điểm lâm sàng là da mỏng, trong suốt, các mảng bầm tím lớn, khuôn mặt đặc trưng (mũi tẹt và mỏng, môi mỏng, tai to, má sâu và các vằn da hổ trên mặt), sự trưởng thành trước tuổi của da.
Các cá thể mắc bệnh có vòng đời ngắn hơn đáng kể (50% tử vong tại tuổi 48) do vỡ đột ngột các cơ quan nội tạng (đại tràng, tử cung) và mạch máu. Hội chứng này ảnh hướng đến toàn bộ hệ mạch (thường là các mạch lớn, trung bình) và tim. Động mạch thường bị ảnh hưởng bao gồm động mạch chủ, thận, mạc treo tràng, chậu, đùi, cũng như động mạch đốt sống và động mạch cảnh (cả đoạn trong sọ và ngoài sọ). Động mạch có thể bị tách mà không có phình từ trước, do đó không thể dự đoán được.
Chẩn đoán hình ảnh không xâm lấn là hướng tiếp cận được lựa chọn để đánh giá các biến đổi về mạch máu. Phȁu thuật chỉ được thực hiện đối với các biến chứng có nguy cơ đe dọa tính mạng, bởi vì tính mỏng manh của mô liên kết, khả năng chảy máu cao, liền vết thương kém làm tăng nguy cơ của phȁu thuật. Theo dõi dài hạn hậu phȁu là bắt buộc. Không có dữ liệu để đưa ra ngưỡng đường kính cần phải can thiệp khi có phình động mạch chủ ngực. Quyết định cần được đưa ra dựa trên từng trường hợp sau khi đã thảo luận đa chuyên khoa.
4. Hội chứng Loeys-Dietz
Là một hội chứng di truyền gen trội trên nhiễm sắc thể thường, được mô tả lần đầu tiên năm 2005, đặc trưng bởi tam chứng phình, xoắn vặn trên suốt chiều dài của cây động mạch; sự cách xa các bộ phận trên cơ thể và lưỡi gà chẻ đôi; cũng như kiểu hình dạng Marfan.
Trong một số dạng, hội chứng Loeys-Dietz chồng lấp đáng kể với Ehlers-Danlos Type
Hội chứng này do đột biến ở cả các gen mã hóa các receptor TGF-ß Type I hoặc Type II (TGFBR1 hoặc TGFBR2). Chỉ số xoắn vặn động mạch sống nền thu được khi chụp MRI ngực có tiêm chất đối quang từ là một chỉ điểm về nguy cơ biến cố tim mạch, không chỉ trong hội chứng Loeys- Dietz mà còn trong các rối loạn mô liên kết khác như hội chứng Marfan và Ehlers-Danlos.
Ở các trẻ có đặc tính sọ mặt càng rõ rệt (chứng liền sớm khớp sọ, thụt hàm, lác ngoài và sụp mí), các đặc điểm lâm sàng nặng (đặc biệt là các tổn thương động mạch chủ) càng rõ rệt . Bởi đặc trưng bệnh lý động mạch lan tỏa và tiến triển nhanh, khuyến cáo phȁu thuật được đưa ra khi đường kính động mạch chủ lên lớn hơn 42 mm.
5. Hội chứng động mạch xoắn vặn (Arterial tortuosity syndrome)
Là bệnh lý rất hiếm gặp, di truyền lặn nhiễm sắc thể thường, đặc trưng bởi sự xoắn vặn, kéo dài, hẹp, phình của các động mạch cỡ trung bình và cỡ lớn.. Hẹp khu trú tại động mạch chủ và động mạch phổi có thể gặp.
Bệnh nhân có khuôn mặt đặc trưng (mặt dài, hẹp khe mi, khe mi mắt xiên xuống, mũi hình mỏ chim, vòm khẩu cái cao và hàm nhỏ), cùng với các dấu hiệu của các bệnh lý mô liên kết nói chung tại da (mềm, dễ kéo dãn), xương (ngón chân nhện, biến dạng lồng ngực, lỏng khớp) chồng lấp với các triệu chứng của hội chứng Marfan.
Tiên lượng bệnh khá tối với tỷ lệ tử vong lên tới 40% vào năm thứ 5 của cuộc đời. Tuy vậy, các nghiên cứu gần đây hơn các gia đình châu Âu cho thấy vȁn có các nhóm bệnh nhân với kiểu hình nhẹ hơn có thể sống sót tới tuổi trưởng thành.
6. Hội chứng phình mạch-viêm xương khớp (AOS: Aortic-osteoarthritis syndrome)
Hội chứng phình mạch – viêm xương khớp là một hội chứng phình tách động mạch chủ ngực mới, chiếm 2% tổng số các trường hợp phình tách ĐM chủ ngực có tính chất gia đình. Bệnh di truyền trội trên nhiễm sắc thể thường do đột biến gen SMAD3 , dȁn đến biểu hiện quá mức các tín hiệu nội tế bào của hệ TGF-0.
Biểu hiện lâm sàng là sự kết hợp của các bất thường khớp xuất hiện sớm (bao gồm viêm xương khớp và viêm xương sụn tách rời), và phình, tách, xoắn vặn động mạch chủ trên toàn bộ chiều dài. Các đặc điểm nhẹ về xương hàm mặt, da, cột sống cũng có thể được thấy, thậm chí bị nhầm lȁn với hội chứng Marfan và Loeys-Dietz.
Chẩn đoán được dựa trên dấu hiệu lâm sàng và sự xác định có đột biến gen SMAD3. Chẹn beta có thể có lợi trong AOS, vì biểu hiện bệnh có các điểm tương tự trong hội chứng Marfan và Loeys-Dietz – những bệnh mà điều trị bằng chẹn beta là tối ưu.
7. Phình, tách động mạch chủ ngực có tính chất gia đình mà không thuộc các hội chứng di truyền
Hầu hết các bệnh nhân có phình, tách động mạch chủ ngực đều không có các hội chứng di truyền được biết trước. Khi nghiên cứu trong gia đình của các bệnh nhân này, có tới 19% những người cận huyết thống (bố mẹ, anh chị em ruột) bị ảnh hưởng. Bệnh di truyền trội trên nhiễm sắc thể thường nhưng có tính đa dạng lâm sàng cao (nhất là ở phụ nữ) và mức độ di truyền đột biến thay đổi theo từng thế hệ. Đột biến các gen đã kể trên như (FBN1, TGFBR1 và TGFBR2) hiếm gặp. Một số đột biến đã được xác định:
- Đột biến gen MYH11 (mã hóa protein myosin chuỗi nặng của tế bào cơ trơn), thường kèm theo còn ống động mạch.
- Đột biến gen ACTA2 (mã hóa alpha-actin đặc hiệu của tế bào cơ trơn) thường kèm theo bệnh lý động mạch vành, đột quỵ hoặc bệnh Moyamoya.
- Đột biến gen MYLK (mã hóa myosin chuỗi nặng).
- Đột biến gen TGFB2 (mã hóa TGF-beta Type 2) thường kèm theo một số đặc điểm da và cột sống thấy trong hội chứng Marfan.
- Đột biến gen PRKG1 (mã hóa PGK I, là một protein phụ thuộc cGMP loại I kiểm soát sự giãn của các tế bào cơ trơn) dȁn tới phình, tách động mạch chủ cấp ở bệnh nhân trẻ tuổi.
Tất cả các trường hợp đã kể trên cho ta thấy mối liên hệ giữa mất chức năng của mô liên kết hoặc giảm tín hiệu TGF-beta hoặc thay đổi chức năng co của cơ trơn với các biểu hiện phình và tách ĐM chủ trên lâm sàng.
Tiên lượng bệnh hiện chưa rõ ràng. Chẩn đoán dựa vào việc xác định các hội chứng gen đã biết, theo sau bởi tham vấn về gen và khảo sát những người cận huyết thống (bố mẹ, anh chị em ruột). Chiến lược quản lý hiện tại là kết hợp giữa chẩn đoán hình ảnh tại thời điểm ban đầu và theo dõi dọc, dựa vào tiền sử gia đình của các biến cố mạch máu.
Leave a Reply