Việc sử dụng SGLT2 inhibitors cùng với sắt tĩnh mạch có thể làm giảm hiệu quả của thuốc sắt. Điều này có thể dẫn đến mức sắt trong cơ thể giảm và gây ra các vấn đề về sức khỏe như thiếu máu. Do đó, trong bài báo “Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure” được đăng trên tạp chí JACC Heart Failure ngày 11/01/2023 sẽ trình bày rõ hơn.
1. Tổng quan
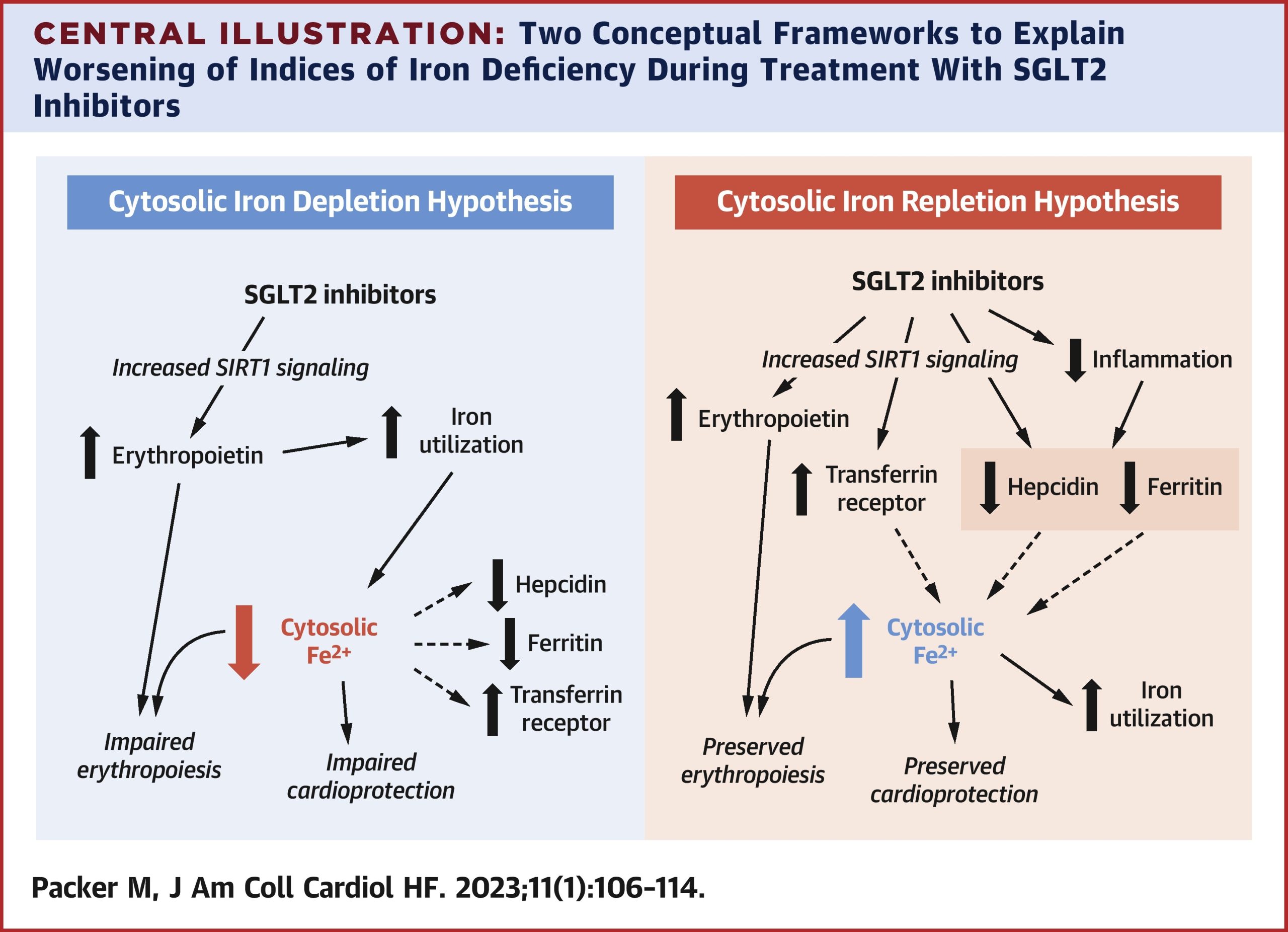
Ở những bệnh nhân bị suy tim, các chất ức chế đồng vận chuyển Natri-Glucose 2 (SGLT2) đã được chứng minh là làm giảm Hepcidin và Ferritin và tăng thụ thể Protein Transferrin, những thay đổi thường làm tình trạng thiếu sắt tuyệt đối trầm trọng hơn, có thể thấy ở bệnh nhân có chế độ ăn uống kém hoặc bệnh đường tiêu hóa, chảy máu, và cả hai đều không bị kích thích bởi thuốc ức chế SGLT2. Do đó, 2 khái niệm khác nhau có thể giải thích tình trạng thay đổi được tìm thấy trong các Protein cân bằng nội môi sắt. Theo “giả thuyết suy giảm sắt tế bào”, tác dụng của thuốc ức chế SGLT2 làm giảm Hepcidin và Ferritin và tăng thụ thể transferrin có liên quan đến sự suy giảm Fe2+ tế bào xảy ra sau khi tăng sử dụng sắt liên quan đến Erythropoietin do thuốc gây ra.
Thuốc bắt chước Erythropoietin (ví dụ, Darbepoietin) gợi ý thiếu sắt, và nó thường đi kèm với tình trạng kháng Erythropoietin được giảm bớt bằng cách bổ sung sắt qua đường tĩnh mạch. Ngược lại, theo “giả thuyết bổ sung sắt trong tế bào”, tác dụng của thuốc ức chế SGLT2 làm giảm Hepcidin và Ferritin và tăng thụ thể Transferrin thể hiện tác dụng trực tiếp của các thuốc này:
- Đảo ngược sự gia tăng Hepcidin và Ferritin liên quan đến viêm. Do đó, giảm bớt các khối chức năng sử dụng sắt.
- Để tăng tín hiệu Sirtuin-1, chất ức chế Hepcidin, đẩy nhanh quá trình phân hủy Ferritin và điều chỉnh tăng Protein thụ thể Transferrin.
Thông qua một trong hai hoặc cả hai cơ chế, việc ức chế trực tiếp Hepcidin và Ferritin sẽ làm tăng Fe2+ tế bào, do đó việc đáp ứng hồng cầu không suy giảm đối với Erythropoietin mà không cần bổ sung sắt qua đường tĩnh mạch.
Toàn bộ bằng chứng lâm sàng ủng hộ “giả thuyết bổ sung sắt trong tế bào” bởi vì các chất ức chế SGLT2 làm tăng hồng cầu và đáp ứng bền vững với Erythropoietin, ngay cả ở những bệnh nhân thiếu sắt quá mức và không điều trị bằng sắt tiêm tĩnh mạch. Do đó, sự xuất hiện của kiểu phản ứng thiếu sắt trong quá trình dùng ức chế SGLT2 không phản ánh tình trạng dự trữ sắt ngày càng xấu đi cần được bổ sung, mà thay vào đó, thể hiện khả năng tình trạng thiếu sắt chức năng liên quan đến viêm nhiễm thường thấy ở bệnh nhân. Đối với suy tim mãn tính, điều trị bằng sắt tiêm tĩnh mạch có thể không cần thiết và có hại về mặt lý thuyết.
2. Giới thiệu
Thuốc ức chế đồng vận chuyển Natri-Glucose 2 (SGLT2) làm giảm nguy cơ tử vong do tim mạch và nhập viện vì suy tim ở bệnh nhân suy tim, cho dù phân suất tống máu giảm hay được bảo tồn. Mặc dù các thuốc này có nghiên cứu chứng minh an toàn cho bệnh nhân, một phân tích gần đây từ nghiên cứ DAPA-HF đã báo cáo rằng việc sử dụng Dapagliflozin ở bệnh nhân suy tim với phân suất tống máu giảm đã dẫn đến tình trạng trầm trọng hơn đáng kể các dấu ấn sinh học của tình trạng thiếu sắt.
Sau 12 tháng điều trị, bệnh nhân ở nhóm Dapagliflozin có khả năng biểu hiện tình trạng thiếu sắt mới khởi phát cao hơn 70% so với nhóm giả dược, được xác định bởi nồng độ Ferritin huyết thanh <100 ng/mL hoặc độ bão hòa Transferrin <20% kết hợp với nồng độ Ferritin huyết thanh <300 ng/mL. Có lẽ quan trọng hơn, những bệnh nhân đã đáp ứng các tiêu chí này khi bắt đầu thử nghiệm cho thấy các chỉ số cân bằng nội môi sắt của họ ngày càng xấu đi sau khi ức chế SGLT2.
Trước những phát hiện này, các tác giả cho rằng những bệnh nhân đang dùng thuốc ức chế SGLT2 có thể được hưởng lợi từ việc bổ sung sắt bổ sung qua đường tĩnh mạch với hy vọng rằng sự kết hợp của 2 phương pháp điều trị có thể mang lại hiệu quả điều trị tổng hợp. Khả năng này dựa trên kết quả của các nghiên cứu ngẫu nhiên có đối chứng với giả dược cho thấy bổ sung sắt qua đường tĩnh mạch (khi không có chất ức chế SGLT2) có thể làm giảm các biến cố suy tim nặng hơn và cải thiện khả năng hoạt động ở bệnh nhân suy tim với phân suất tống máu giảm đáp ứng các tiêu chí hiện tại về điều trị suy tim.
Đáng chú ý là gần 50% bệnh nhân trong thử nghiệm DAPA-HF được coi là thiếu sắt khi bắt đầu thử nghiệm, và thêm 15% trong nhóm thuần tập ban đầu trở nên thiếu sắt mới khi điều trị bằng Dapagliflozin. Vì vậy, nếu sử dụng các phương pháp đánh giá tình trạng thiếu sắt thông thường, gần 2/3 bệnh nhân suy tim với phân suất tống máu giảm sẽ được coi là bị thiếu sắt và được cho liệu pháp bổ sung sắt sau vài tháng điều trị bằng thuốc ức chế SGLT2, gợi ý rằng liệu pháp phối hợp có thể trở thành một giải pháp hữu hiệu.
3. Điều gì làm thay đổi tình trạng của dấu ấn sinh học trong tình trạng thiếu sắt trong quá trình sử dụng ức chế SGLT2?
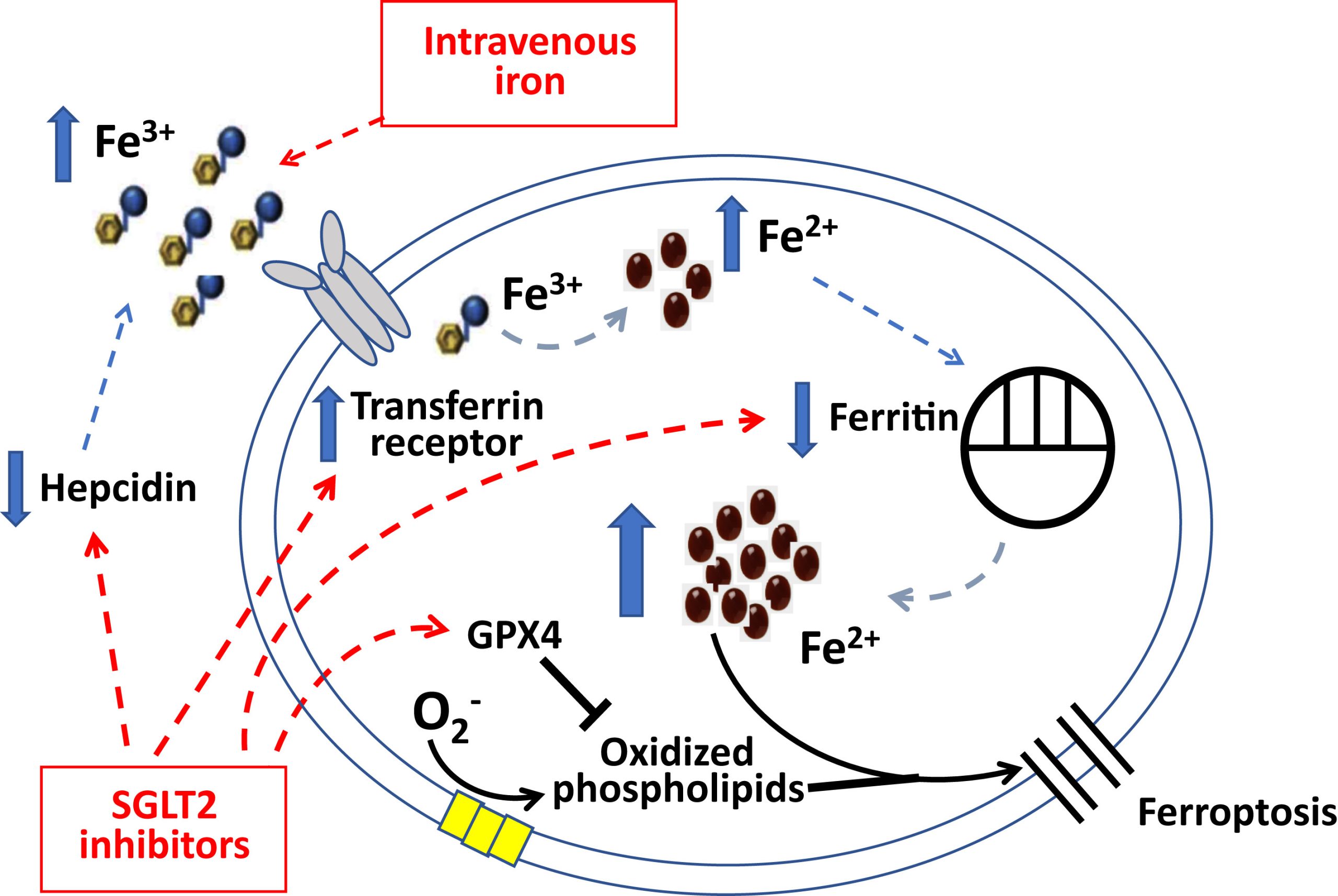
Sắt cần thiết cho quá trình tổng hợp heme và các cụm sắt-lưu huỳnh trong ty thể của tiền chất hồng cầu (dẫn đến tổng hợp huyết sắc tố) và trong tế bào cơ tim (dẫn đến sản xuất adenosine triphosphate [ATP]). Sắt chịu trách nhiệm trực tiếp cho các hiệu ứng này là nhóm tế bào chứa ion sắt có khả năng phản ứng cao (Fe2+) liên kết yếu, chiếm 5% – 10% lượng sắt dự trữ trong tế bào.
Điều gì quyết định kích thước của nhóm sắt Fe2+ tham gia phản ứng sinh học? Sắt đi vào máu dưới dạng sắt (Fe3+), một quá trình được điều hòa bởi Hepcidin (ngăn chặn sự hấp thu sắt từ đường tiêu hóa và giải phóng sắt từ các đại thực bào tham gia vào quá trình tái tạo hồng cầu già). Sắt Fe3+ lưu thông được vận chuyển vào tế bào khi Protein vận chuyển của nó (transferrin) gắn với Protein thụ thể 1 vận chuyển , và phức hợp này được nội hóa. Quá trình khử Fe3+ giải phóng Fe2+ vào nhóm tế bào chất phản ứng sinh học, nhưng kích thước của nhóm này được điều chỉnh chặt chẽ, và hầu hết sắt nội bào được cô lập dưới dạng Fe3+ không hoạt động trong lồng nano Ferritin, giúp giải phóng sắt vào tế bào chất khi cần thiết. Khi dự trữ sắt trong cơ thể bị cạn kiệt (như trong tình trạng thiếu dinh dưỡng hoặc xuất huyết tiêu hóa), việc điều chỉnh giảm Hepcidin và điều chỉnh tăng Protein thụ thể transferrin 1 phối hợp với nhau để tạo điều kiện thuận lợi cho việc hấp thu và đưa sắt vào các tế bào sản xuất Heme, và, đồng thời Ferritin bị phân hủy, có tác dụng giải phóng sắt cô lập từ các vị trí dự trữ nội bào. Nồng độ Hepcidin và Ferritin thấp và nồng độ cao của Protein thụ thể Transferrin 1 (thường được đo bằng thụ thể Transferrin hòa tan) trong máu lưu thông thường được coi là là dấu hiệu của tình trạng thiếu sắt. Tuy nhiên, kiểu thay đổi đặc biệt này trong các Protein cân bằng nội môi sắt không phải là thước đo kích thước của nhóm Fe2+ trong tế bào chất; thay vào đó, những thay đổi này phản ánh các phản ứng bù trừ khi Fe2+ trong tế bào cạn kiệt và cần được bổ sung.
Nguồn: Milton Packer, Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure, JACC Heart Failure, 2023.
4. Giải thích tình trạng xấu đi của các dấu ấn sinh học sắt trong quá trình điều trị với thuốc ức chế SGLT2.
Mặc dù sự thay đổi về chỉ dấu sinh học sắt này thường cho thấy sự cạn kiệt nguồn dự trữ sắt của cơ thể do thiếu hụt dinh dưỡng hoặc xuất huyết tiêu hóa, trạng thái sinh lý bệnh khác cũng dẫn đến một tình trạng đặc biệt là giảm Hepcidin và Ferritin cùng với sự gia tăng thụ thể Transferrin hòa tan.
4.1. Tăng sinh hồng cầu
Kích thích tạo hồng cầu (bất kể nguyên nhân nào) tiêu thụ Fe2+ tế bào trong quá trình hình thành heme, và kết quả là giảm Fe2+, tế bào kích hoạt làm giảm Hepcidin và Ferritin và tăng thụ thể Transferrin hòa tan, như một phản ứng thích nghi để bổ sung khả năng chứa sắt. Hoạt động phối hợp của các protein này có tác dụng duy trì đủ lượng Fe2+ trong tế bào để đảm bảo rằng quá trình tạo hồng cầu hiệu quả có thể được duy trì. Các chất bắt chước Erythropoietin (ví dụ, darbepoietin) gợi ra một kiểu phản ứng thiếu sắt, có khả năng phản ánh sự giảm đáng kể về mặt lâm sàng của Fe2+ trong tế bào vì:
- Bệnh nhân với kiểu này cho thấy phản ứng kém với Erythropoietin.
- Bổ sung Fe2+ trong tế bào bằng việc bổ sung sắt qua đường tĩnh mạch giúp khắc phục tình trạng kháng Erythropoietin.
4.2. Viêm hệ thống
Ở những bệnh được đặc trưng bởi tình trạng viêm mãn tính, hoạt hóa cytokine kích thích trực tiếp quá trình tổng hợp Hepcidin và Ferritin. Tác động của viêm đối với Hepcidin và Ferritin làm chậm lượng sắt có thể được hấp thụ từ tá tràng hoặc giải phóng từ đại thực bào cũng như lượng sắt nội bào có thể được giải phóng khỏi quá trình cô lập Ferritin. Kết quả là tình trạng thiếu Fe2+ tế bào, mặc dù lượng dự trữ Fe3+ toàn thân và nội bào không bị cạn kiệt, tức là tình trạng thiếu sắt tế bào mang tính chức năng, chứ không phải tuyệt đối.
Tình trạng thiếu sắt chức năng có thể đáp ứng tốt với việc cung cấp sắt qua đường tĩnh mạch (chứ không phải đường uống). Tuy nhiên, nếu tình trạng viêm mãn tính có thể được cải thiện, kích thích sinh học đối với quá trình tổng hợp Hepcidin và Ferritin giảm xuống, dẫn đến sự gia tăng phản ứng sinh học trong tế bào. Fe2+, mặc dù nồng độ Hepcidin và Ferritin trong tuần hoàn đang giảm. Trong những trường hợp này, sự xuất hiện của kiểu phản ứng thiếu sắt không phản ánh sự hiện diện của tình trạng thiếu sắt, mà thay vào đó, phản ánh sự thuyên giảm của tình trạng viêm nhiễm liên quan đến khối chức năng về sử dụng sắt. Sự đảo ngược tình trạng thiếu sắt chức năng qua trung gian viêm tạo ra những thay đổi trong các dấu ấn sinh học sắt lưu hành gần giống với sự phát triển của tình trạng thiếu sắt tuyệt đối. Tuy nhiên, do nồng độ Fe2+ trong tế bào được hỗ trợ và tăng cường, nên việc thiếu sắt chức năng không đi kèm với kháng Erythropoietin và không cần bổ sung sắt để duy trì hồng cầu.
Cơ chế nào trong 2 cơ chế này có thể giải thích những thay đổi tìm thấy được trong dấu ấn sinh học thiếu sắt trong quá trình điều trị bằng thuốc ức chế SGLT2 ở bệnh nhân suy tim? Trong các thử nghiệm đối chứng với giả dược, chất ức chế SGLT2 đã được chứng minh là làm tăng thụ thể Erythropoietin và Transferrin hòa tan trong khi làm giảm Hepcidin và Ferritin. Những thay đổi như vậy tương ứng với một trong 2 khái niệm được mô tả. Tuy nhiên, khái niệm thứ nhất được khẳng định dựa trên giả định rằng nồng độ Fe2+ trong tế bào bị cạn kiệt do quá trình tạo hồng cầu tăng cường, trong khi khái niệm thứ hai giả định rằng nồng độ Fe2+ trong tế bào đang được bổ sung do giảm viêm hoặc bởi các tác động khác giảm Stress tế bào. Khái niệm đầu tiên dẫn đến kết luận rằng việc bổ sung sắt là rất quan trọng để mang lại kết quả tối ưu cho bệnh nhân, trong khi khái niệm thứ hai gợi ý rằng việc bổ sung sắt là không cần thiết. Khái niệm nào giải thích tác dụng của thuốc ức chế SGLT2?
4.3. Các cơ chế tế bào ảnh hưởng đến cân bằng nội môi sắt trong quá trình ức chế SGLT2.
Theo “giả thuyết suy giảm sắt tế bào”, chất ức chế SGLT2 trực tiếp kích thích sản xuất Erythropoietin, có lẽ thông qua khả năng điều chỉnh tăng tín hiệu sirtuin-1 (SIRT1) đã được thiết lập của chúng. SIRT1 tăng cường sản xuất Erythropoietin thông qua tác dụng của nó để kích hoạt yếu tố cảm ứng thiếu oxy-2α. Hoạt động của Erythropoietin để kích thích quá trình tạo hồng cầu làm cạn kiệt nhóm Fe2+ trong tế bào, do đó kích hoạt sự ức chế Hepcidin và Ferritin và điều chỉnh tăng Protein thụ thể transferrin như một phản ứng bù trừ. Tuy nhiên, những thay đổi mang tính phản ứng này không thể phục hồi Fe2+ trong tế bào nếu nguồn thực phẩm và/hoặc dự trữ sắt ở đại thực bào và gan là quá ít. Sự cạn kiệt Fe2+ trong tế bào làm suy yếu đáp ứng của hồng cầu với Erythropoietin, dẫn đến tình trạng kháng Erythropoietin phụ thuộc sắt, giống như tình trạng đã thấy khi sử dụng các chất kích thích tạo Erythropoietin.
Theo “giả thuyết bổ sung sắt trong tế bào” , việc điều chỉnh tăng tín hiệu SIRT1 không chỉ thúc đẩy quá trình tổng hợp Erythropoietin mà còn trực tiếp làm giảm Hepcidin và Ferritin và tăng protein thụ thể transferrin, không phụ thuộc vào quá trình tạo hồng cầu. Các chất ức chế SGLT2 phát huy tác dụng chống viêm quan trọng (có thể qua trung gian SIRT1) do đó đảo ngược tác dụng kích thích đối với cả Hepcidin và Ferritin. SIRT1 ức chế quá trình tổng hợp Hepcidin ở cả tế bào gan và đại thực bào; SIRT1 kích hoạt hệ số điều hòa Gamma-1 Alpha được kích hoạt bởi chất tăng sinh Peroxisome, hoạt động để điều chỉnh tăng protein thụ thể transferrin; và kích thích yếu tố gây thiếu Oxy-2 alpha bởi SIRT1 làm tăng biểu hiện của chất đồng hoạt hóa thụ thể hạt nhân, thúc đẩy quá trình ăn Ferritino, do đó làm thoái hóa Ferritin. Do đó, thông qua tác dụng chống viêm và các hoạt động tế bào khác ức chế Hepcidin và Ferritin (không phụ thuộc vào quá trình tạo hồng cầu), các chất ức chế SGLT2 có thể trực tiếp vượt qua các khối chức năng bên trong do sử dụng sắt gây ra do viêm hệ thống. Bởi vì dự trữ sắt trong tế bào không bị cạn kiệt ở những bệnh nhân bị thiếu sắt chức năng, nên việc đảo ngược các khối chức năng khi sử dụng sắt với việc sử dụng chất ức chế SGLT2 có tác dụng làm tăng Fe2+ tế bào, do đó cho phép đáp ứng hồng cầu không bị suy giảm, trong trường hợp không bổ sung sắt.
4.4. Bằng chứng lâm sàng ủng hộ giả thuyết bổ sung sắt tế bào
Để xác định khái niệm nào có thể áp dụng được trong quá trình điều trị bằng thuốc ức chế SGLT2, sẽ là lý tưởng nếu các nhà lâm sàng có thể đo lượng Fe2+ trong tế bào, nhưng điều đó là không thể trong môi trường lâm sàng. Tuy nhiên, 2 bằng chứng từ các nghiên cứu lâm sàng ủng hộ giá trị của “giả thuyết bổ sung sắt trong tế bào”.
Đầu tiên, tình trạng thiếu sắt gặp ở 50% bệnh nhân suy tim với phân suất tống máu giảm không phải do tình trạng thiếu hụt tuyệt đối liên quan đến chế độ ăn uống kém hoặc xuất huyết tiêu hóa. Thay vào đó, tỷ lệ thiếu sắt cao là do sự cản trở chức năng giải phóng sắt từ các kho dự trữ hiện có. Bệnh nhân suy tim có tình trạng viêm kích thích sản xuất cả Hepcidin và Ferritin, giải thích tại sao nồng độ Hepcidin trong huyết thanh lại cao tăng trong suy tim song song với việc kích hoạt các con đường tiền viêm. Tương tự như vậy, bệnh nhân suy tim có nồng độ Ferritin tăng lên và mức độ gia tăng này rõ rệt đến mức sự hiện diện của suy tim làm thay đổi ngưỡng của Ferritin được sử dụng để chẩn đoán tình trạng thiếu sắt. Sự suy giảm sắt ở bệnh nhân không bị suy tim thường được xác định khi Ferritin huyết thanh giảm xuống <30 ng/mL. Tuy nhiên, ngưỡng này tăng gấp 10 lần ở bệnh nhân suy tim, những người có thể được chẩn đoán là thiếu sắt với nồng độ ferritin cao tới 299 ng/mL (hoặc thậm chí cao hơn). Mặc dù một số nghiên cứu đã báo cáo tình trạng thiếu sắt tuyệt đối ở bệnh nhân suy tim, những nghiên cứu này đã đánh giá các đoàn hệ nhỏ được lựa chọn kỹ càng đã được kiểm tra tủy xương, điều này rất khó giải thích. Mặc dù có thể một số bệnh nhân bị suy tim tiến triển đã cạn kiệt nguồn dự trữ sắt, nhưng hầu hết bệnh nhân bị suy tim có các triệu chứng từ nhẹ đến trung bình (như trường hợp trong nghiên cứu DAPA-HF) và có tình trạng thiếu sắt chức năng.
Thứ hai, nếu những thay đổi về dấu ấn sinh học sắt trong quá trình sử dụng ức chế SGLT2 thể hiện tình trạng thiếu sắt ngày càng trầm trọng hơn, thì những bệnh nhân vốn đã thiếu sắt trước khi điều trị sẽ có phản ứng suy giảm đối với các loại thuốc này, liên quan đến tác dụng của chúng đối với cả xương, tuỷ và tim. Cụ thể, những bệnh nhân thiếu sắt sẽ cho thấy tạo hồng cầu kém vì sự cạn kiệt Fe2+ trong tế bào dẫn đến kháng Erythropoietin. Hơn nữa, ATP bị cạn kiệt ở bệnh nhân suy tim, đặc biệt khi bệnh nhân thiếu sắt, phục hồi ATP là cơ sở cho việc sử dụng các chất bổ sung sắt đường tĩnh mạch để giảm nguy cơ các biến cố suy tim nghiêm trọng. Do đó, nếu các chất ức chế SGLT2 làm giảm Fe2+ tế bào, chúng sẽ làm trầm trọng thêm sự suy giảm ATP liên quan đến thiếu sắt trong cơ tim, dẫn đến tăng nguy cơ biến cố suy tim và hạn chế khả năng giảm nhập viện do suy tim của thuốc ức chế SGLT2. Tuy nhiên, trong nghiên cứu DAPA-HF, khi so sánh với những bệnh nhân có đủ sắt, những bệnh nhân thiếu sắt lúc ban đầu cho thấy lượng huyết sắc tố tăng tương tự và giảm tương tự nguy cơ biến cố suy tim khi đáp ứng với Dapagliflozin, mặc dù các dấu ấn sinh học của tình trạng thiếu sắt trở nên xấu đi ở những bệnh nhân này trong quá trình điều trị. Điều quan trọng là mức độ tăng hồng cầu được tạo ra bởi các chất ức chế SGLT2 không suy giảm theo thời gian, phát hiện sẽ khó giải thích nếu bệnh nhân thực sự trở nên thiếu sắt ngày càng nhiều hơn.
Cần lưu ý rằng—trước khi điều trị bằng thuốc ức chế SGLT2—nhiều bệnh nhân trong nghiên cứu DAPA-HF được coi là thiếu sắt cũng bị thiếu máu, ngụ ý rằng mức Fe2+ tế bào trong tiền chất hồng cầu không đủ để duy trì đủ lượng hồng cầu. Tuy nhiên, sau khi điều trị bằng thuốc ức chế SGLT2, những bệnh nhân bị thiếu máu do thiếu sắt có khả năng điều chỉnh tình trạng thiếu máu, mặc dù <1% được bổ sung sắt qua đường tĩnh mạch. Chuỗi sự kiện này chỉ có thể xảy ra nếu việc ức chế SGLT2 có tác dụng làm giảm bớt mà không làm trầm trọng thêm tình trạng nồng độ Fe2+ trong tế bào thấp có tác dụng hạn chế tạo hồng cầu ở những bệnh nhân thiếu máu này. Sự giảm thiểu như vậy phù hợp với “giả thuyết bổ sung sắt tế bào”, nhưng nó không phù hợp với định nghĩa của “giả thuyết suy giảm sắt tế bào”.
Nguồn: Milton Packer, Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure, JACC Heart Failure, 2023.
Cần lưu ý rằng sự phát triển của thuốc ức chế SGLT2 và thuốc bổ sung sắt tiêm tĩnh mạch cho bệnh suy tim đã diễn ra song song và các nghiên cứu quy mô lớn với một loại thuốc thường không thu nhận bệnh nhân dùng thuốc kia. Do đó, các bác sĩ không biết liệu thuốc ức chế sắt và thuốc ức chế SGLT2 tiêm tĩnh mạch có tương tác với nhau hay không nếu được kê đơn cùng nhau. Việc sử dụng đồng thời các loại thuốc này trong thực hành lâm sàng đặc biệt có khả năng xảy ra vì thuốc ức chế SGLT2 làm giảm độ bão hòa ferritin và transferrin huyết thanh, và do đó, chúng làm tăng khả năng bệnh nhân bị chẩn đoán là thiếu sắt, và do đó, được cân nhắc điều trị bằng sắt tiêm tĩnh mạch.
5. Tổng kết
Bằng chứng sẵn có cho thấy rằng:
- Hầu hết bệnh nhân suy tim nhẹ đến trung bình đều có tình trạng viêm thúc đẩy sự phát triển của tình trạng thiếu sắt chức năng.
- Việc sử dụng các chất ức chế SGLT2 dường như làm giảm bớt tình trạng thiếu sắt chức năng song song với tác dụng chống viêm của chúng mà không cần sử dụng sắt tiêm tĩnh mạch.
- Thuốc ức chế SGLT2 có tác dụng làm trầm trọng thêm các dấu ấn sinh học tuần hoàn thường được sử dụng để đánh giá tình trạng sắt ở bệnh nhân suy tim, nhưng những thay đổi này của các dấu ấn sinh học sắt không xác định được tình trạng thiếu sắt nội bào làm hạn chế tác dụng có lợi của các thuốc này trong cơ thể.
Trên thực tế, việc làm giảm tình trạng thiếu sắt chức năng trong quá trình sử dụng ức chế SGLT2 sẽ làm giảm bớt, thay vì tăng thêm, nhu cầu bổ sung sắt qua đường tĩnh mạch ở những bệnh nhân này. Do đó, trước khi sắt tiêm tĩnh mạch được kê đơn rộng rãi cho bệnh nhân suy tim có biểu hiện thiếu sắt trong khi dùng thuốc ức chế SGLT2, cần các nghiên cứu tính hữu ích của liệu pháp phối hợp để xác định xem “giả thuyết bổ sung sắt trong tế bào” hay “giả thuyết về cạn kiệt sắt trong tế bào”. Hiệu quả và độ an toàn của việc sử dụng đồng thời thuốc ức chế SGLT2 và sắt tiêm tĩnh mạch ở bệnh nhân suy tim nên được kiểm tra trước khi khuyến cáo điều trị kết hợp.
Nguồn: Milton Packer, Potential Interactions When Prescribing SGLT2 Inhibitors and Intravenous Iron in Combination in Heart Failure, JACC Heart Failure, 2023 Jan, 11 (1) 106–114.
Leave a Reply